Prof. Dr.
Josef Ecker, born in 1978, is a distinguished biologist specializing in lipid
metabolism and its implications for health and disease. He completed his
biology studies at the University of Regensburg, earning his doctorate in 2007.
Following his Ph.D., he conducted research at the University of California,
Berkeley, focusing on lipid metabolism.
Currently,
Prof. Ecker leads the Functional Lipidomics and Metabolism Research group at
the Institute of Clinical Chemistry and Laboratory Medicine, University
Hospital Regensburg. His research centers on the interplay between dietary
fats, gut microbiota, and systemic lipid metabolism, particularly concerning
intestinal lipid absorption and its role in diseases such as cancer.
In addition
to his role in Regensburg, Prof. Ecker is a principal investigator in the Collaborative
Research Center (CRC) 1371 at the Technical University of Munich (TUM). Within
this consortium, he co-leads Project P13, which investigates the impact of
dietary fat and gut microbiota interactions on intestinal lipid absorption,
systemic lipid metabolism, and intestinal cancer.
Prof. Ecker
has an extensive publication record, with his work being cited over 3,500
times, reflecting his significant contributions to the field of lipidomics.
His
research aims to elucidate the molecular mechanisms underlying lipid metabolism
and its influence on health, providing insights that could lead to novel
therapeutic strategies for metabolic diseases and cancer. Many thanks, Josef,
for your great presentation!
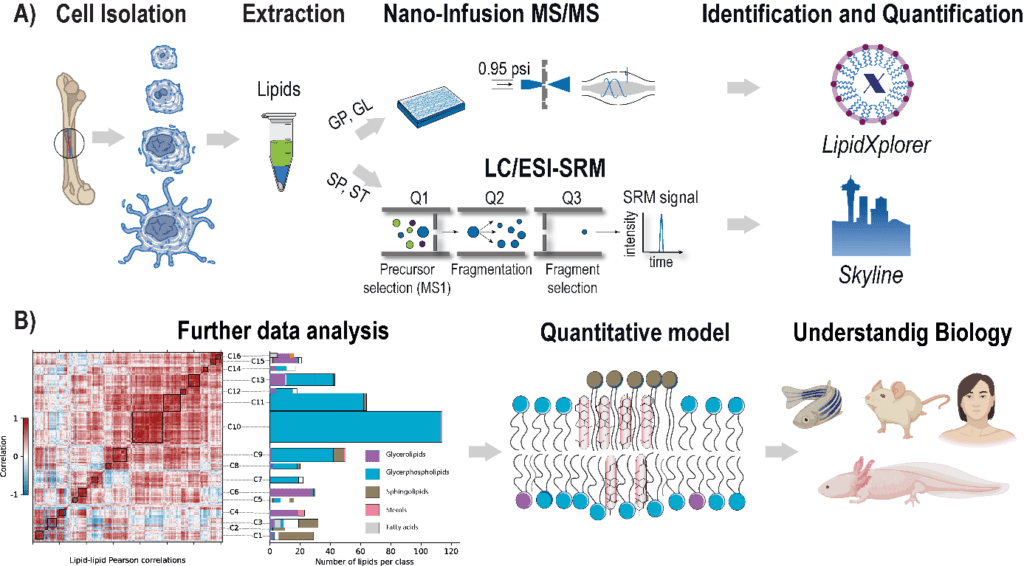







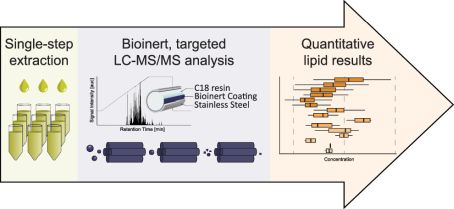
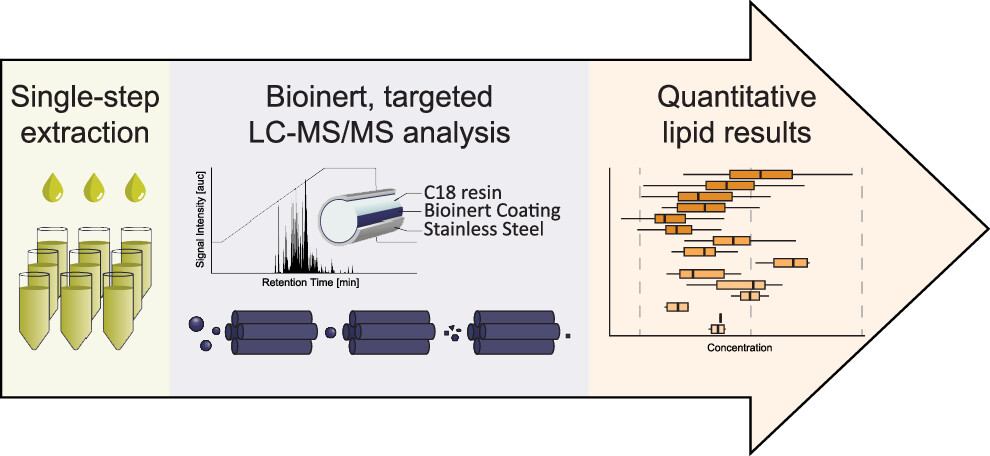

 The rising prevalence of obesity globally presents a critical social challenge, threatening to reverse the progress made in life expectancy in developed nations. Gaining a thorough understanding of the mechanisms behind fat cell production is, thus, essential for the development of novel treatment strategies. While past studies focused on individual molecular layers, we recognize the importance of considering higher network connectivity levels, particularly lipid feedback controlling the master regulator of adipogenesis, PPARG. Our approach integrates advanced lipidomics and proteomics techniques, monitoring PPARG and the lipidome during perturbations in vitro. Through innovative multiomics strategies and high-throughput imaging, we dissect feedback networks and identify lipid regulators of PPARG. This research not only advances scientific knowledge but also holds promise for future obesity drug development, potentially revolutionizing treatment approaches.
The rising prevalence of obesity globally presents a critical social challenge, threatening to reverse the progress made in life expectancy in developed nations. Gaining a thorough understanding of the mechanisms behind fat cell production is, thus, essential for the development of novel treatment strategies. While past studies focused on individual molecular layers, we recognize the importance of considering higher network connectivity levels, particularly lipid feedback controlling the master regulator of adipogenesis, PPARG. Our approach integrates advanced lipidomics and proteomics techniques, monitoring PPARG and the lipidome during perturbations in vitro. Through innovative multiomics strategies and high-throughput imaging, we dissect feedback networks and identify lipid regulators of PPARG. This research not only advances scientific knowledge but also holds promise for future obesity drug development, potentially revolutionizing treatment approaches. 


{kind=link}
{kind=link}
{kind=link}
{kind=link}