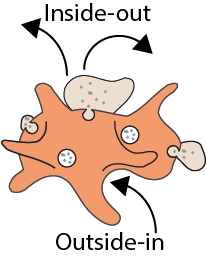
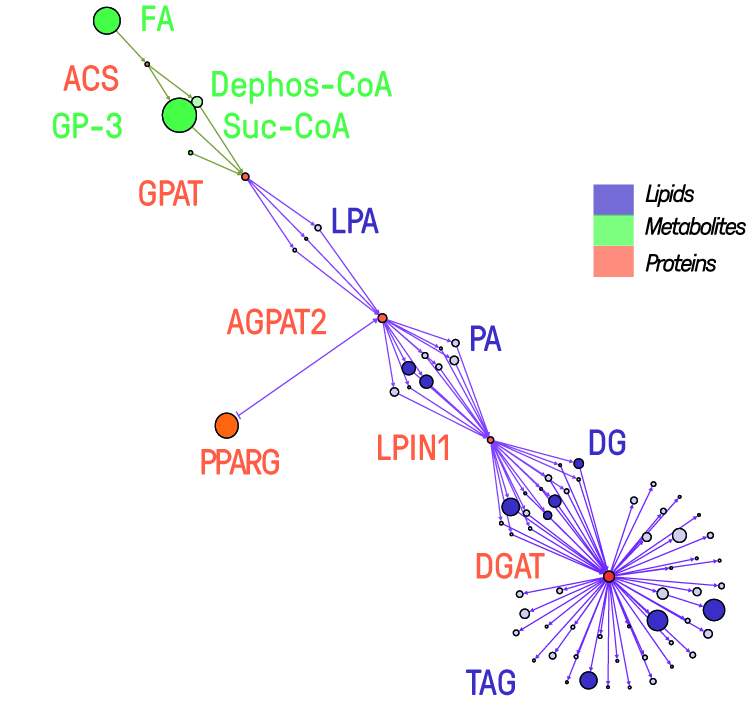
The rising prevalence of obesity globally presents a critical social challenge, threatening to reverse the progress made in life expectancy in developed nations. Gaining a thorough understanding of the mechanisms behind fat cell production is, thus, essential for the development of novel treatment strategies. While past studies focused on individual molecular layers, we recognize the importance of considering higher network connectivity levels, particularly lipid feedback controlling the master regulator of adipogenesis, PPARG. Our approach integrates advanced lipidomics and proteomics techniques, monitoring PPARG and the lipidome during perturbations in vitro. Through innovative multiomics strategies and high-throughput imaging, we dissect feedback networks and identify lipid regulators of PPARG. This research not only advances scientific knowledge but also holds promise for future obesity drug development, potentially revolutionizing treatment approaches.
Lipidomics encompasses analytical approaches that aim to identify and quantify the complete set of lipids, defined as lipidome in a given cell, tissue, or organism, and their interactions with other molecules. Most lipidomics workflows are based on mass spectrometry and have been proven a powerful tool in system biology in concert with other Omics disciplines. Unfortunately, computational approaches for this relatively young discipline are limited and only accessible to some specialists. Search engines, quantification algorithms, visualization tools, and databases developed by the ‘Lipidomics Informatics for Life-Science’ (LIFS) initiative will provide a structured and standardized format for broad access to these specialized bioinformatics pipelines. Many medical challenges related to lipid metabolic alterations will be highly supported by such capacity-building. Within LIFS, we already provide access to several tools, workflows, tutorials, and training via a unified web portal (https://lifs-tools.org/).
Platelet integrity and function critically depend on lipid composition. However, the lipid inventory in platelets was hitherto not quantified (Peng et al., Blood, 2018). Today our lab examines the lipidome of murine and human platelets using lipid-category tailored protocols on quantitative lipidomics platforms. We commonly can cover the platelet lipidome, which is comprised out of 500 lipids (99.9% of the total lipid mass) over a concentration range of seven orders of magnitude. We conduct systematic comparison of lipidomics network in resting and activated murine platelets, validated in human platelets, where we inter alia revealed that less than 20% of the platelet lipidome is changed upon activation, involving mainly lipids containing arachidonic acid. However, the most interesting work that we currently conducting in close collaboration with our partners is the analysis of different diseases models (Scheller et al.,Haematologica, 2019) which display and thrombotic phenotype. E.g., Sphingomyelin phosphodiesterase-1 (Smpd1) deficiency results in a very specific modulation of the platelet lipidome (Peng et al., Blood, 2018) with an order of magnitude up-regulation of lyso-sphingomyelin (SPC), and subsequent modification of platelet activation and thrombus formation, which sheds light on novel mechanisms important for platelet function, and has therefore the potential to open novel diagnostic and therapeutic opportunities.
Metabolic dysfunctions are not only highly correlated with insulin resistance and diabetes, but are also associated with a 10 fold higher risk to develop Alzheimer’s disease. The proposed project joins forces between two Leibniz institutions with different expertise to establish a unique research platform for Translational Neuroscience. Here, we will break ground by introducing lipidomics to the field of synapse biology, by investigating insulin resistance in conjunction with high Abetaload and by studying the effect whether synaptic disease states result in an altered lipid composition that in turn leads to synaptic dysfunction in brain.
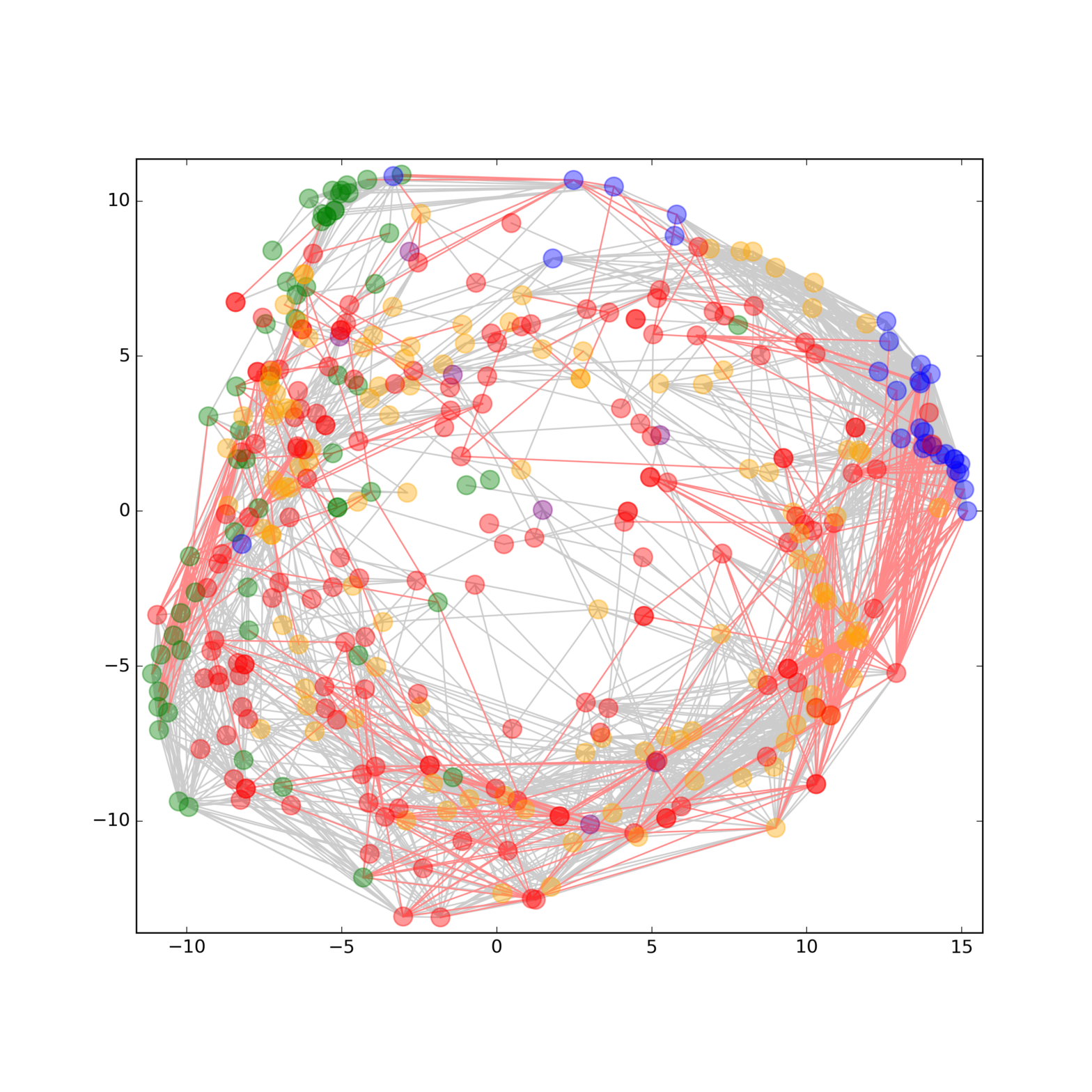
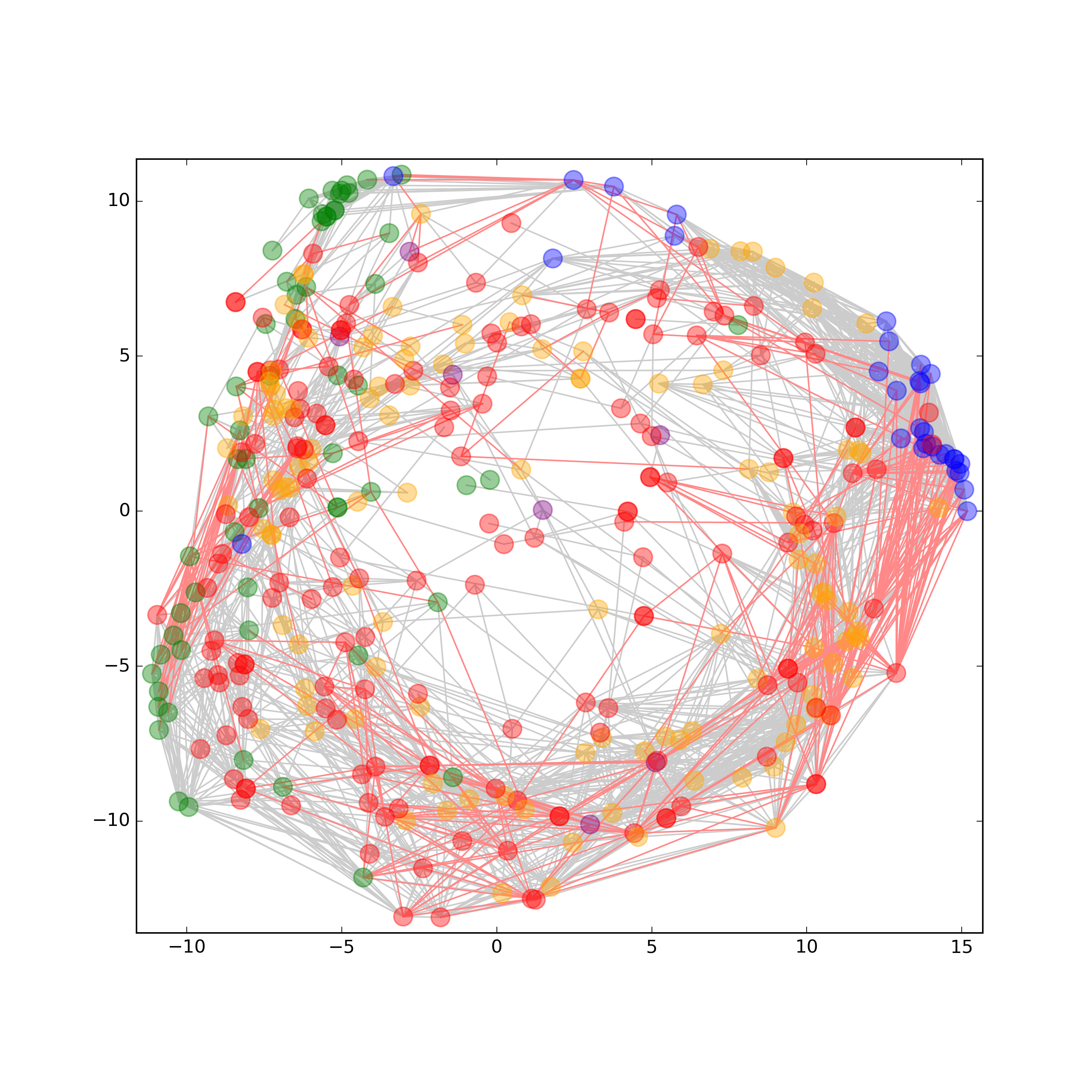
Phenotypes at cellular and organism level are a result of a multitude of different molecular species. Thereby, interconnected networks are at the heart of both signaling pathways and complex traits that mediate adaptive plasticity and determine phenotypes. To answer the question how different molecular layers are connected and to gain deeper insights into the underlying mechanisms that determine a certain phenotype, a comprehensive and representative analysis of the molecular species involved is necessary [2]. Historically, each molecule class (e.g. DNA, RNA, proteins, metabolites, and lipids) has been studied separately in large scale omics experiments to look for relationships within biological processes. Using this strategy, we have assembled some of the molecular pieces related to signaling networks, but many interactions between them are still unrevealed or unexplained due to the restrictive single‐data‐type study designs. Therefore, multimolecular approaches on the sample processing as well as on the data analysis side are a prerequisite to obtain an integrated perspective. Read more in our recent publications SIMPLEX a multiomics for systems biology (https://www.ncbi.nlm.nih.gov/pubmed/26814187).
 The rising prevalence of obesity globally presents a critical social challenge, threatening to reverse the progress made in life expectancy in developed nations. Gaining a thorough understanding of the mechanisms behind fat cell production is, thus, essential for the development of novel treatment strategies. While past studies focused on individual molecular layers, we recognize the importance of considering higher network connectivity levels, particularly lipid feedback controlling the master regulator of adipogenesis, PPARG. Our approach integrates advanced lipidomics and proteomics techniques, monitoring PPARG and the lipidome during perturbations in vitro. Through innovative multiomics strategies and high-throughput imaging, we dissect feedback networks and identify lipid regulators of PPARG. This research not only advances scientific knowledge but also holds promise for future obesity drug development, potentially revolutionizing treatment approaches.
The rising prevalence of obesity globally presents a critical social challenge, threatening to reverse the progress made in life expectancy in developed nations. Gaining a thorough understanding of the mechanisms behind fat cell production is, thus, essential for the development of novel treatment strategies. While past studies focused on individual molecular layers, we recognize the importance of considering higher network connectivity levels, particularly lipid feedback controlling the master regulator of adipogenesis, PPARG. Our approach integrates advanced lipidomics and proteomics techniques, monitoring PPARG and the lipidome during perturbations in vitro. Through innovative multiomics strategies and high-throughput imaging, we dissect feedback networks and identify lipid regulators of PPARG. This research not only advances scientific knowledge but also holds promise for future obesity drug development, potentially revolutionizing treatment approaches.