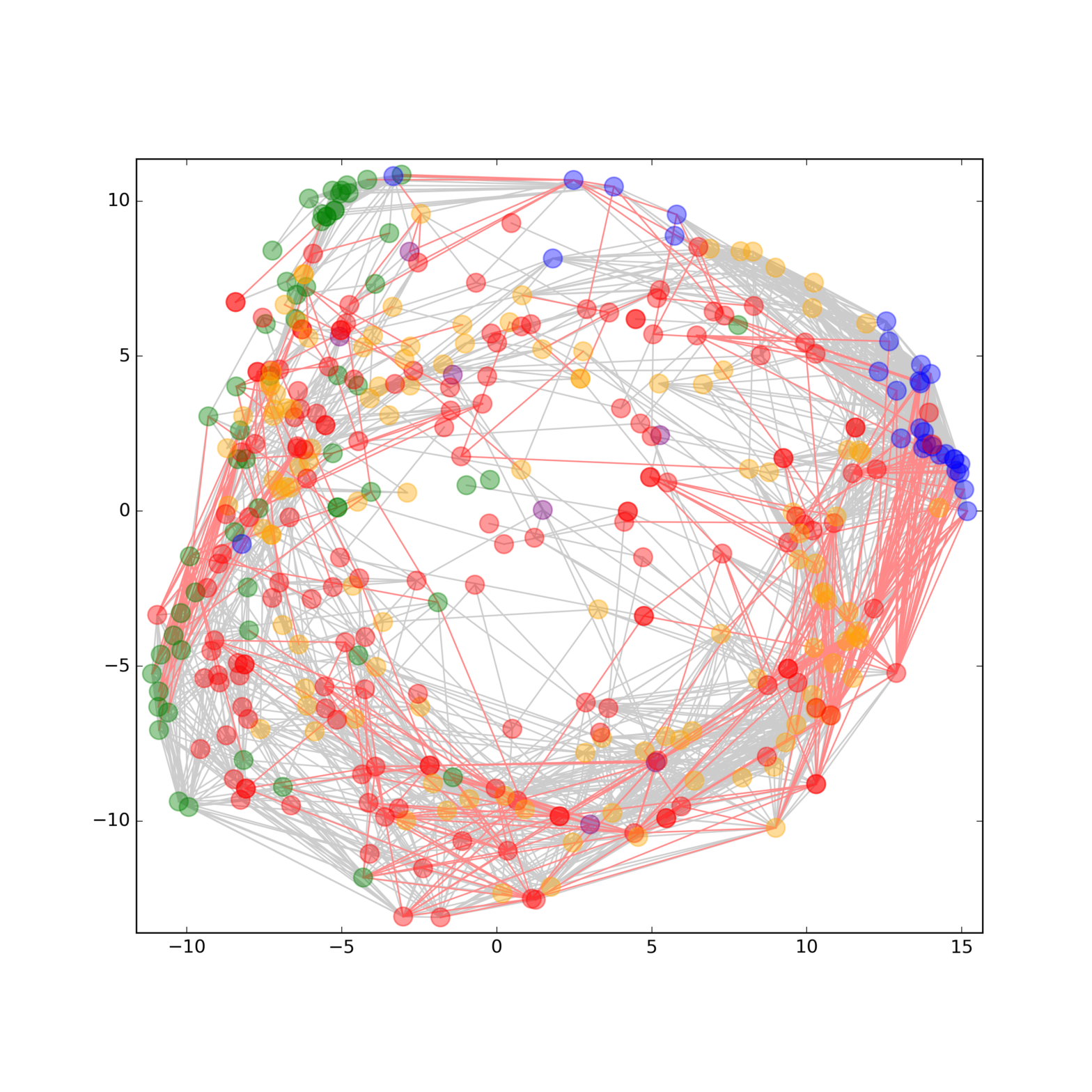
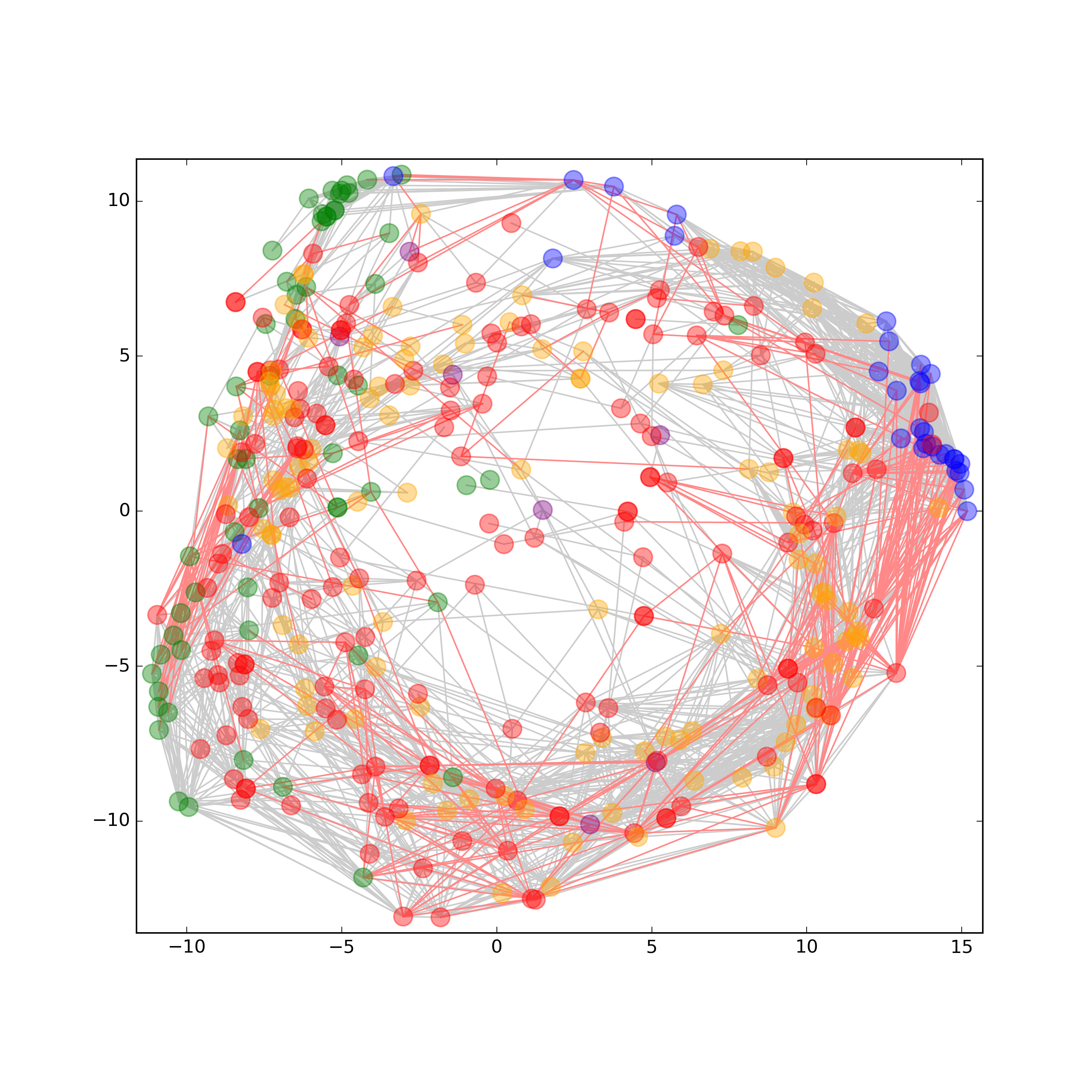
Phenotypes at cellular and organism level are a result of a multitude of different molecular species. Thereby, interconnected networks are at the heart of both signaling pathways and complex traits that mediate adaptive plasticity and determine phenotypes. To answer the question how different molecular layers are connected and to gain deeper insights into the underlying mechanisms that determine a certain phenotype, a comprehensive and representative analysis of the molecular species involved is necessary [2]. Historically, each molecule class (e.g. DNA, RNA, proteins, metabolites, and lipids) has been studied separately in large scale omics experiments to look for relationships within biological processes. Using this strategy, we have assembled some of the molecular pieces related to signaling networks, but many interactions between them are still unrevealed or unexplained due to the restrictive single‐data‐type study designs. Therefore, multimolecular approaches on the sample processing as well as on the data analysis side are a prerequisite to obtain an integrated perspective. Read more in our recent publications SIMPLEX a multiomics for systems biology (https://www.ncbi.nlm.nih.gov/pubmed/26814187).
Phenotypes at cellular and organism level are a result of a multitude of different molecular species. Thereby, interconnected networks are at the heart of both signaling pathways and complex traits that mediate adaptive plasticity and determine phenotypes. To answer the question how different molecular layers are connected and to gain deeper insights into the underlying mechanisms that determine a certain phenotype, a comprehensive and representative analysis of the molecular species involved is necessary [2]. Historically, each molecule class (e.g. DNA, RNA, proteins, metabolites, and lipids) has been studied separately in large scale omics experiments to look for relationships within biological processes. Using this strategy, we have assembled some of the molecular pieces related to signaling networks, but many interactions between them are still unrevealed or unexplained due to the restrictive single‐data‐type study designs. Therefore, multimolecular approaches on the sample processing as well as on the data analysis side are a prerequisite to obtain an integrated perspective. Read more in our recent publications SIMPLEX a multiomics for systems biology (https://www.ncbi.nlm.nih.gov/pubmed/26814187).
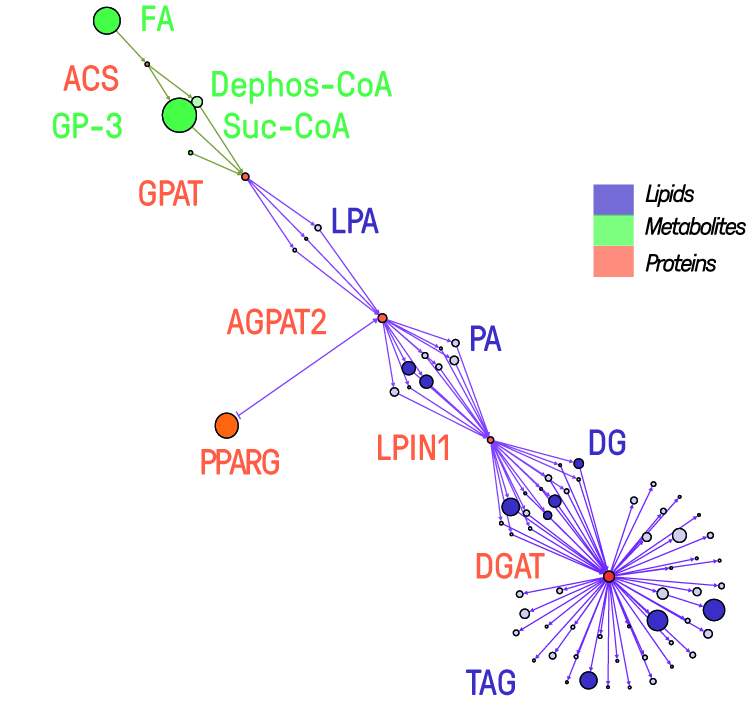
 Lipidomics encompasses analytical approaches that aim to identify and quantify the complete set of lipids, defined as lipidome in a given cell, tissue, or organism, and their interactions with other molecules. Most lipidomics workflows are based on mass spectrometry and have been proven a powerful tool in system biology in concert with other Omics disciplines. Unfortunately, computational approaches for this relatively young discipline are limited and only accessible to some specialists. Search engines, quantification algorithms, visualization tools, and databases developed by the ‘Lipidomics Informatics for Life-Science’ (LIFS) initiative will provide a structured and standardized format for broad access to these specialized bioinformatics pipelines. Many medical challenges related to lipid metabolic alterations will be highly supported by such capacity-building. Within LIFS, we already provide access to several tools, workflows, tutorials, and training via a unified web portal (https://lifs-tools.org/).
Lipidomics encompasses analytical approaches that aim to identify and quantify the complete set of lipids, defined as lipidome in a given cell, tissue, or organism, and their interactions with other molecules. Most lipidomics workflows are based on mass spectrometry and have been proven a powerful tool in system biology in concert with other Omics disciplines. Unfortunately, computational approaches for this relatively young discipline are limited and only accessible to some specialists. Search engines, quantification algorithms, visualization tools, and databases developed by the ‘Lipidomics Informatics for Life-Science’ (LIFS) initiative will provide a structured and standardized format for broad access to these specialized bioinformatics pipelines. Many medical challenges related to lipid metabolic alterations will be highly supported by such capacity-building. Within LIFS, we already provide access to several tools, workflows, tutorials, and training via a unified web portal (https://lifs-tools.org/).